[性能評価] GPUを用いたLAMMPS分子動力学シミュレーション
はじめに
プロメテック・ソフトウェアの三上です。HPCインフラエンジニアとしてHPC基盤の設計や構築に携わっています。
GPUは流体シミュレーションなど高速化の効果が高いと評価されていますが、今回、GPUを用いて分子動力学(MD)シミュレーションがどの程度高速化できるのかを評価してみました。MDシミュレーションは多数の分子のポテンシャルや座標を計算する必要があり、GPUの効果が期待される分野の一つです。MDシミュレーション領域では多くのOSS(オープンソースソフトウェア)が利用できますが、今回はその一つであるLAMMPSを評価しています。
LAMMPSのインストールや利用方法はLAMMPSのウェブサイト上に掲載されています。LAMMPSの性能評価結果は以下に掲載されていますが、古いデータが多く最近のデータセンター向けGPUのNVIDIA A100のベンチマークデータがありません。そのため、NVIDIA A100を用いてベンチマークを取得してみました。
汎用MD計算ソフト LAMMPSについて
LAMMPSはサンディア国立研究所により開発された古典分子動力学(MD)計算プログラムです。金属、半導体、生体分子、ポリマー、メゾスコピック系物質など多様な物質に対してのポテンシャルが用意されているのが特徴です。
今回の評価環境
- LAMMPS version: 2Aug2023
- CUDA Version: 12.2
- GPU: NVIDIA A100 80GB PCIe
- CPU: Intel(R) Xeon(R) Gold 5317 CPU @ 3.00GHz
LAMMPSのビルド
LAMMPSのビルドには、CMakeまたはMakeを用いますが今回はCMakeを用いました。CMakeに必要なCMakeLists.txtはLAMMPSのパッケージに同梱されています。
cmake ../cmake -C ../cmake/presets/basic.cmake \
-D PKG_ASPHERE=ON \
-D PKG_GPU=ON \
-D GPU_API=cuda \
-D GPU_ARCH=sm_80 \
-DBIN2C=/usr/local/cuda/bin/bin2c
Examples
LAMMPSを用いて、いくつかの入力ファイルに対して実行結果をCPUとGPUを用いて測定して比較してみました。
今回使用したスクリプトは、LAMMPS公式ページのものを引用しています。
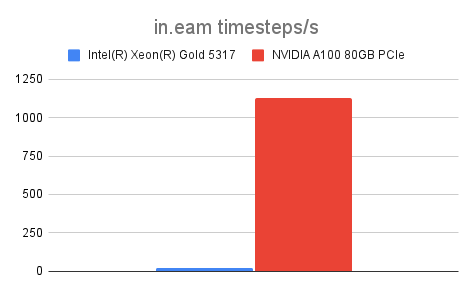
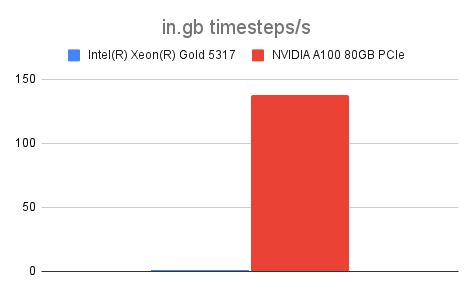
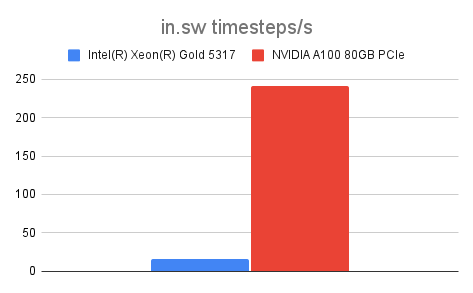
性能の結果は以下のようになりました。スクリプト詳細については、以下に示しています。
| CPU[timesteps/s] | GPU[timesteps/s] | 高速化倍率 | |
|---|---|---|---|
| in.eam | 20.565 | 1127.763 | 54.84 |
| in.gb | 1.235 | 137.812 | 111.59 |
| in.sw | 16.348 | 241.385 | 14.77 |
EAM Potential
今回使用したLAMMPSスクリプトは以下です。
# bulk Cu lattice
log logs/log.eam append
variable x index 1
variable y index 1
variable z index 1
variable xx equal 20*$x
variable yy equal 20*$y
variable zz equal 20*$z
units metal
atom_style atomic
lattice fcc 3.615
region box block 0 ${xx} 0 ${yy} 0 ${zz}
create_box 1 box
create_atoms 1 box
pair_style eam
pair_coeff 1 1 Cu_u3.eam
velocity all create 1600.0 376847 loop geom
neighbor 1.0 bin
neigh_modify every 1 delay 5 check yes
fix 1 all nve
timestep 0.005
thermo 10
# dump mydmp all atom 100 dump.eam.lammpstrj
run 10000以下のように、GPUパッケージを用いてNVIDIA A100上で計算を行う場合とCPU(Intel(R) Xeon(R) Gold 5317 CPU @3.00GHz)1コアで計算を行う場合の二通りで計算を行いました。
$ lmp -sf gpu -pk gpu 1 -in in.eam // GPUを用いる場合
$ lmp -in in.eam // CPUを用いる場合GB Potential
今回使用したLAMMPSスクリプトは以下です。
# Gay-Berne benchmark
# biaxial ellipsoid mesogens in isotropic phase
# shape: 2 1.5 1
# cutoff 4.0 with skin 0.8
# NPT, T=2.4, P=8.0
units lj
atom_style ellipsoid
# creation
#lattice sc 0.22
#region box block 0 32 0 32 0 32
#create_box 1 box
#create_atoms 1 box
#set group all quat/random 982381
read_data data.gb
compute rot all temp/asphere
group spheroid type 1
variable dof equal count(spheroid)+3
compute_modify rot extra ${dof}
velocity all create 2.4 41787 loop geom
pair_style gayberne 1.0 3.0 1.0 4.0
pair_coeff 1 1 1.0 1.0 1.0 0.5 0.2 1.0 0.5 0.2
neighbor 0.8 bin
timestep 0.002
thermo 10
dump mydmp all atom 5 dump.eam2.lammpstrj
# equilibration
#fix 1 all npt/asphere temp 2.4 2.4 0.1 iso 5.0 8.0 0.1
#compute_modify 1_temp extra ${dof}
#run 100
#write_restart tmp.restart
fix 1 all npt/asphere temp 2.4 2.4 0.2 iso 8.0 8.0 0.2
run 1000以下のように、GPUパッケージを用いてNVIDIA A100上で計算を行う場合とCPU(Intel(R) Xeon(R) Gold 5317 CPU @3.00GHz)1コアで計算を行う場合の二通りで計算を行いました。
$ lmp -sf gpu -pk gpu 1 -in in.gb // GPUを用いる場合
$ lmp -in in.gb // CPUを用いる場合SW Potential
今回使用したLAMMPSスクリプトは以下です。
# bulk Si via Stillinger-Weber
units metal
atom_style atomic
lattice diamond 5.431
region box block 0 20 0 20 0 10
create_box 1 box
create_atoms 1 box
pair_style sw
pair_coeff * * Si.sw Si
mass 1 28.06
velocity all create 1000.0 376847 loop geom
neighbor 1.0 bin
neigh_modify delay 5 every 1
fix 1 all nve
timestep 0.001
thermo 10
dump mydmp all atom 5 dump.sw.lammpstrj
run 20000以下のように、GPUパッケージを用いてNVIDIA A100上で計算を行う場合とCPU(Intel(R) Xeon(R) Gold 5317 CPU @3.00GHz)1コアで計算を行う場合の二通りで計算を行いました。
$ lmp -sf gpu -pk gpu 1 -in in.sw // GPUを用いる場合
$ lmp -in in.sw // CPUを用いる場合まとめ
GPUを用いたLAMMPS分子動力学シミュレーションの性能評価結果を紹介しました。
プロメテックグループ最大規模のカンファレンスPrometech Simulation Conference 2023では、「GPUを活かした計算化学と機械学習」と題して、DFT計算から得られた学習データからニューラルネットワークを用いて機械学習を行い、ポテンシャル関数を生成してシミュレーションを行う例でGPUの性能評価を紹介しますので、是非そちらもご覧ください。
